

Akromegalia – mało znana choroba dużych ludzi
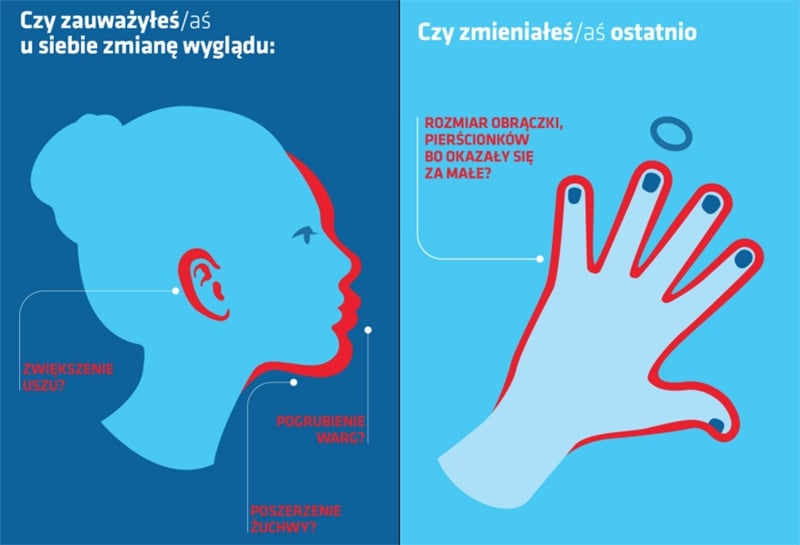
Chociaż są już dorośli, ich ciało zaczyna się powiększać. Rosną im stopy, dłonie, uszy i nos. Z czasem zaczynają wyróżniać się swoim wyglądem - ich postura jest większa od przeciętnej, a nienaturalnie zaostrzona żuchwa sprawia, że twarz nabiera groźnego wyrazu.
To, co brzmi jak scenariusz do filmu science fiction, to realia życia osób dotkniętych rzadką chorobą o podłożu hormonalnym – akromegalią. Maj to miesiąc budowania wiedzy na temat tej nietypowej i trudnej w rozpoznaniu choroby.
Szacuje się, że na akromegalię choruje około 70 osób na milion. Pierwszymi, zauważalnymi objawami u chorych są powiększone ręce i stopy. Mogą również wystąpić takie symptomy, jak powiększenie czoła, szczęki i nosa. Choroba ta związana jest z obecnością łagodnego guza przysadki mózgowej (gruczolaka przysadki). To właśnie obecność guza wywołuje nadmierne wydzielanie hormonu wzrostu. W rezultacie u pacjentów dochodzi m.in. do przerostu tkanek miękkich. Ich narządy wewnętrzne – serce, tarczyca, wątroba, a nawet jelita mogą być większe, niż u przeciętnego człowieka.
Akromegalia to nie tylko zmiany w wyglądzie
Nadmiar hormonu wzrostu powoduje, że chorzy na akromegalią mają bardzo charakterystyczną aparycję. Jedną z najbardziej znanych osób z akromegalią był amerykański aktor Rondo Hatton – jego postura z powodzeniem pozwalała mu na liczne występy w filmach grozy. Nietypowy wygląd spowodowany akromegalią otworzył też drzwi do kariery Mauricie Tillet’owi, pierwowzorowi bajkowego Shreka, który w latach 40. ubiegłego wieku był jednym z najbardziej popularnych zawodników wrestlingu.
Jednak Rondo Hattona, jak i Mauricie Tilletta, obok popularności, jaka przyszła wraz z pojawieniem się choroby, łączyło coś jeszcze – obaj zmarli z powodu ataku serca niedługo po swoich 50. urodzinach. Jak podkreślają eksperci, śmierć z powodu powikłań sercowych jest jedną z częstszych przyczyn zgonów w przypadku postępującej, nieleczonej akromegalii.
– Powiększenie serca, wątroby oraz przerost innych organów, to tylko niektóre z powikłań, jakie wiążą się z tą chorobą. Rozrost tych narządów powoduje z kolei inne, groźne schorzenia. Chorzy obok deformacji kości często zmagają się z nadciśnieniem tętniczym czy niebezpiecznymi w skutkach zaburzeniami układu krążenia. Zmiany te można jednak spowolnić lub zatrzymać dzięki możliwościom, jakie daje współczesna medycyna. Gdyby Rondo Hatton czy Mauricie Tillet byli właściwie leczeni, prawdopodobnie żyliby znacznie dłużej. Dziś nie mamy problemów z leczeniem. W Polsce nie odbiega ono od standardów światowych, trudności sprawia natomiast opóźniona diagnostyka – mówi prof. Wojciech Zgliczyński, kierownik Kliniki Endokrynologii CMKP w Szpitalu Bielańskim w Warszawie.
Zbyt późne rozpoznanie
Po rozpoznaniu choroby, najważniejsze jest szybkie wdrożenie odpowiedniej terapii. Podstawową metodą leczenia jest operacyjne usunięcie guza. Operacja jest możliwa najczęściej na wczesnym etapie choroby, gdy guz jest niewielki. W przeciwnym razie konieczna staje się farmakoterapia. Podstawę leczenia farmakologicznego stanowią analogi somatostatyny, które skutecznie obniżają stężenie hormonu wzrostu i zmniejszają objętość guza. Leki te stosowane są z sukcesem także w leczeniu przedoperacyjnym, w celu zmniejsza wielkości zmiany nowotworowej. Eksperci zauważają, że o ile szybkie wdrożenie leczenia może zatrzymać dalszy rozwój choroby, nie cofnie zmian, do których zdążyło już dojść, dlatego tak niezbędna jest szybka diagnostyka. Wczesne zdiagnozowanie akromegalii zwiększa skuteczność leczenia, wpływa na zmniejszenie kosztów opieki systemowej nad chorym i zwiększa szansę na poprawienie długości i jakości życia pacjentów. Tymczasem w Polsce rozpoznanie akromegalii zajmuje średnio aż 10 lat i jak podkreślają eksperci, często jest przypadkowe.
– Proces przemiany jest stopniowy, nie dzieje się z dnia na dzień, dlatego też ani osoba, której choroba dotyczy, ani jej najbliżsi mogą nie dostrzegać symptomów choroby. Bywa, że przemianę u chorych zauważają dopiero ich dawno niewidziani znajomi, szczególnie jeśli chory podczas ostatniego spotkania miał inne rysy twarzy. Czasami to ich sugestie doprowadzają do poszukiwania przyczyny zachodzących zmian. Sami pacjenci często bagatelizują pierwsze objawy, nie szukają przyczyny powiększających się dłoni, mimo że są przez to zmuszeni do zrezygnowania z noszenia biżuterii lub do powiększenia obrączki. Mężczyźni potrafią tłumaczyć sobie, że przerost dłoni spowodowany jest ciężką pracą fizyczną. Co ciekawe, chociaż choroba dotyczy obu płci, częściej rozpoznawana jest wśród kobiet, ponieważ z natury przywiązują one większą uwagę do wyglądu – zaznacza prof. Wojciech Zgliczyński.
Na wydłużoną diagnostykę nie bez wpływu pozostaje również rzadkość występowania akromegalii oraz niewielka świadomość społeczeństwa na temat tej choroby. Na jakie symptomy warto więc zwrócić uwagę?
– Najbardziej widocznymi zmianami są te w wyglądzie fizycznym, a więc powiększone dłonie, stopy, rozrośnięta żuchwa oraz nos i uszy. Z czasem jednak u chorych pojawia się także ogólne zmęczenie, osłabienie. Charakterystycznym objawem akromegalii jest też wzmożona potliwość. Pacjentom dokuczają również dolegliwości ze strony układu kostno-stawowego w postaci zniekształceń, zmian zwyrodnieniowych i bólów kostno-stawowych. Ważnym symptomem, szczególnie u chorych z dużym guzem przysadki, jest ból głowy i zaburzenia widzenia wywołane uciskiem guza na nerw wzrokowy oraz powikłania sercowo-naczyniowe, arytmie i nadciśnienie. Jeśli więc zauważymy u siebie tego typu symptomy, którym towarzyszą m.in. zmiany w wyglądzie, należy jak najszybciej zgłosić się do lekarza. Akromegalię można leczyć, a powikłania zatrzymać, jednak do tego niezbędne jest szybkie wdrożenie leczenia. Nieleczona akromegalia prowadzi do skrócenia życia chorych nawet o 30 proc. w porównaniu z populacją ogólną. To mnóstwo czasu, który ktoś mógłby zyskać, gdyby tylko udało się szybko rozpoznać chorobę – podkreśla prof. Wojciech Zgliczyński.
Obecnie w Polsce żyje około 2,5 tys. chorych na akromegalię. Co roku rozpoznaje się blisko 200 nowych przypadków tej choroby.
pr
Data publikacji: 29.05.2019 r.




